Performs molecular Monte Carlo simulation of single chain protein or single chain nucleic acids.
The Monomer Monte Carlo module is accessible from the Simulate section of the main menu.
The purpose of the module is perform a molecular simulation of an input single chain protein or single chain nucleic acid by sampling backbone torsion angles.
The starting structure must be a complete structure without missing residues. Atom and residue naming must be compatiable with those defined in the CHARMM force field See Notes on Starting Structures and Force Fields and PDB Scan for further details.
Only single chain protein or single chain nucleic acids are supported. Systems with multiple chains can be modeled using Complex Monte Carlo.
The output file format is DCD since in most cases many structures are generated. There is no option to save the output files in PDB format. One can use Extract Utilities to convert DCD files to multi-frame PDB files.
Structures are generated by Markov Monte Carlo sampling of backbone torsion angles. Energetics of torsion angles are determined using CHARMM force field parameters.
Typically, between 10,000 to 50,000 structures are required to sample adequate configuration space for most problems.
Parameters are supplied to help guide the Monte Carlo sampling such as temperature, control of single move angle sampling per region, and directed Monte Carlo options to guide the radius of gyration (Rg) to a user supplied value.
A utility is provided to overlap accepted structures onto a single reference frame. This is useful to visualize relative configuration coverage in an ensemble.
Several options are offered to check for atomic overlap: heavy atoms, all, backbone, and atom name. If one chooses the atom name option, then the user will be prompted to supply an atom name that should exist in all residues and a overlap distance cutoff value. Other options set the cutoff distance automatically.
In Advanced Input options are provided to reject structures based on Rg value, position of atoms in the Z-direction and via atomic constraints provided as a list in a text file as described in Constraints. These options are not mutually exclusive and can be used in the same run as needed.
Typical workflows involve generating an ensemble of structures using this module, then energy minimizing the ensemble using Energy Minimization, then calculating scattering from the ensemble using modules in Calculate, and finaly comparing results to experimental data using modules in Analyze.
In many situations, multiple runs need to be carried out to find structures that cover configuration space and have scattering profiles that are in agreement with experimental data. One can use Merge Utilties to combine both the structures (DCD files) and SAS profiles into a single new DCD and single directory will correctly numbered SAS profiles.
To simulate long random coil regions, usually at the ends of globular proteins, it is often neccessary to sub-sample accepted structures as adjacent structures can be correlated. To obtain adequate power-law scaling, one can sub-sample a trajectory using Extract Utitilies using the periodic sampling option.
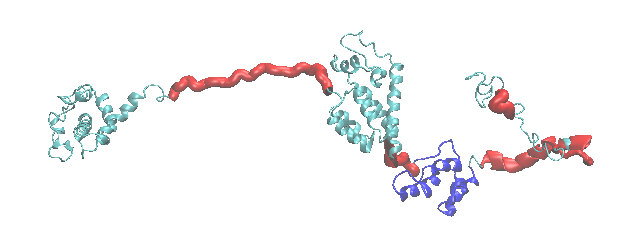
This example generates a series of structures to sample configurations of the HIV-1 Gag protein. The cartoon of the starting structure highlights the flexible regions (red) and structure alignment region (blue).
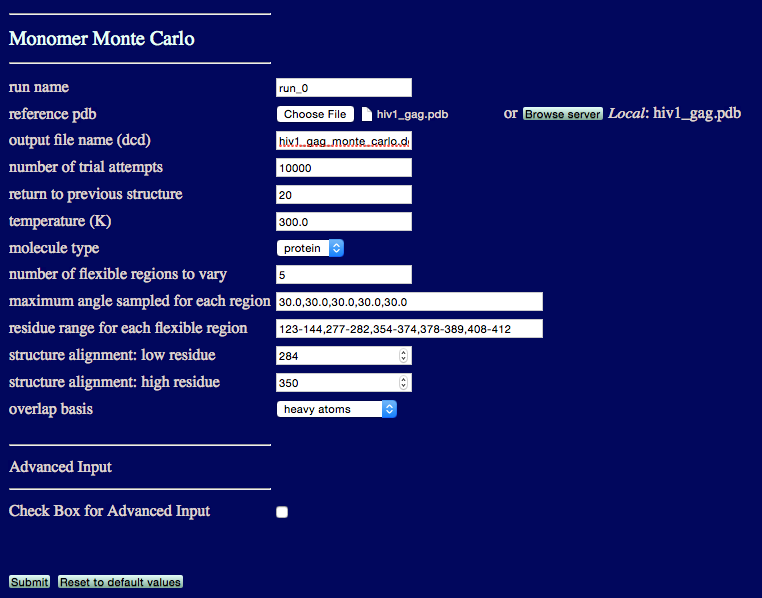
run name user defined name of folder that will contain the results.
reference pdb PDB file with naming information and coordinates of the starting structure.
output file name Name of ouput DCD file containing accepted structures from the simulation.
number of trial attempts Number of Monte Carlo moves to attempt.
return to previous structure After this number of Monte Carlo moves fails to find an accepted configuration, re-load a previously accepted structure.
temperature (K) Simulation temperature.
molecule type Select either protein or RNA.
number of flexible regions to vary An integer value indicating the number of regions to sample backbone torsions.
maximum angle sampled for each region Angle, in degrees, that can be sampled in a single move for each region.
residue range for each flexible region Residue numbers defining each flexible region.
structure alignment: low residue Residue to define the beginning of region used to align all structures.
structure alignment: high residue Residue to define the end of region used to align all structures.
overlap basis Select either heavy atoms, all, backbone or enter atom name. The atom name option will spawn futher inputs:
overlap basis Enter an atom name to check for overlap.
overlap cutoff (angsgtroms) Overlap basis atoms closer than this distance defines an overlap condition.
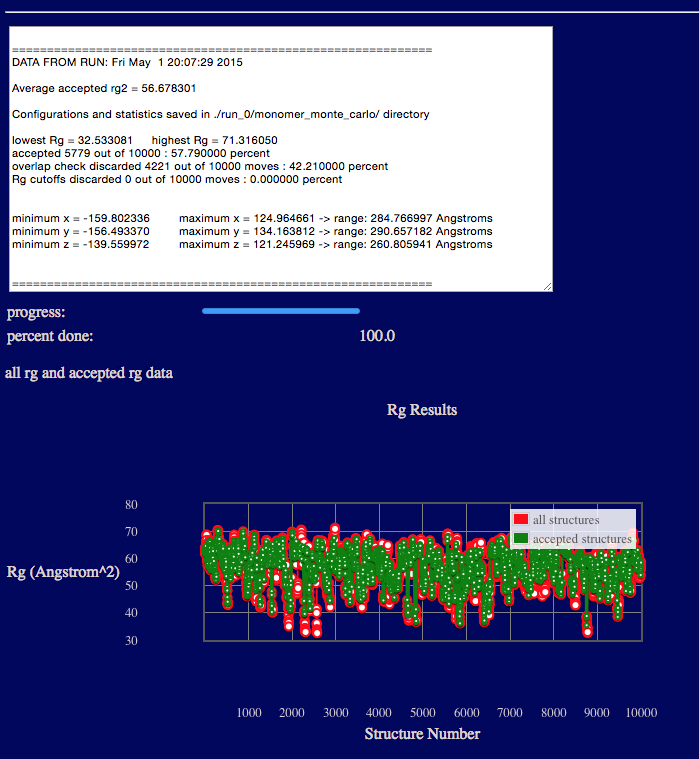
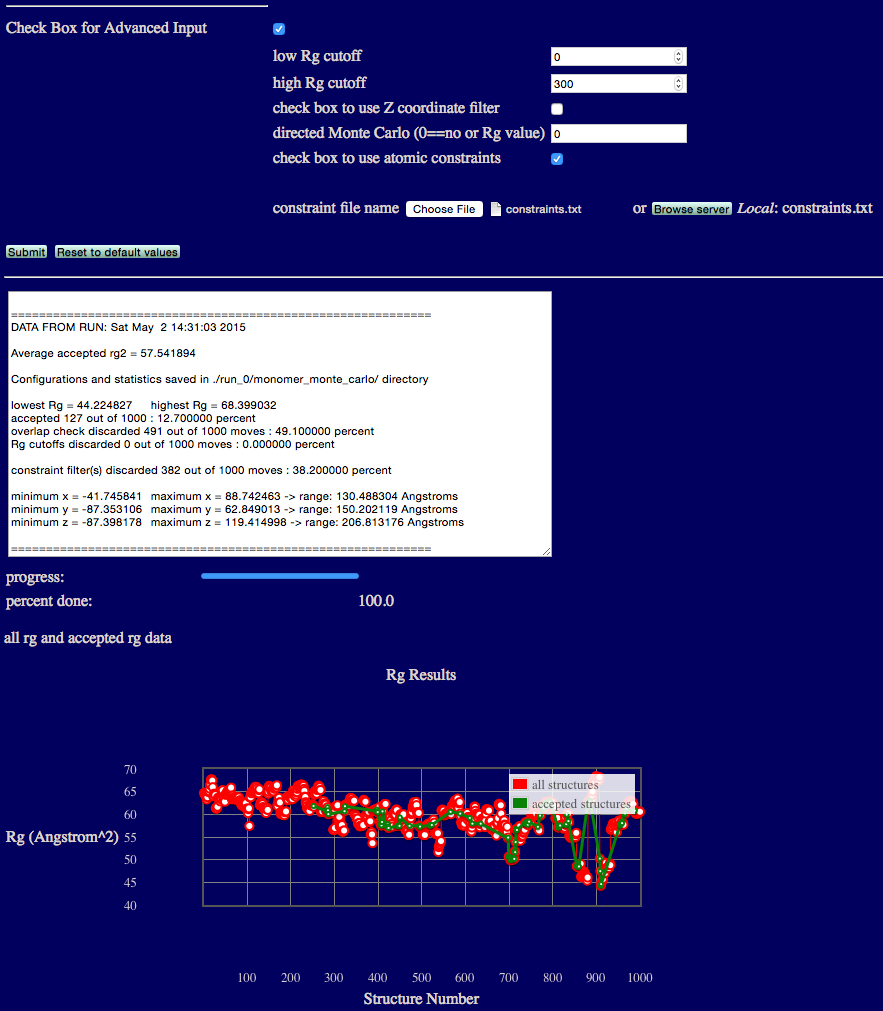
The output will indicate various Rg values from the ensemble, acceptance and overlap statistics, and dimensions of the accepted structures in the final ensemble.
Results are written to a new directory within the given "run name" as noted in the output. In addition, a plot of Rg versus structure number is shown.
Several files are generated and saved to the "run name" monomer_monte_carlo directory. A copy of the original input PDB file, the output DCD file containing accepted structures, files with Rg values as shown in the plot on the web-page, and run statistics.
./run_0/monomer_monte_carlo/hiv1_gag.pdb
./run_0/monomer_monte_carlo/hiv1_gag_monte_carlo.dcd
./run_0/monomer_monte_carlo/hiv1_gag_monte_carlo.dcd.all_rg_results_data.txt
./run_0/monomer_monte_carlo/hiv1_gag_monte_carlo.dcd.accepted_rg_results_data.txt
./run_0/monomer_monte_carlo/hiv1_gag_monte_carlo.dcd.stats
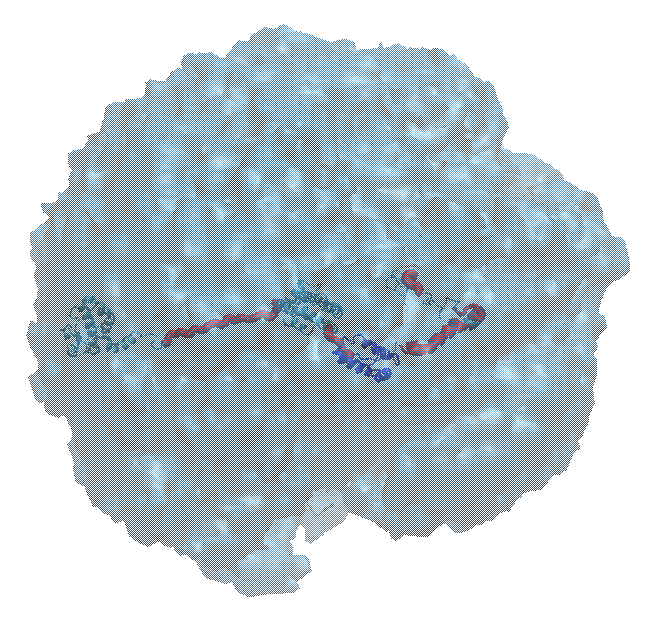
In the figure below, the original input structure of hiv1_gag inside the envelope sampled by all accepted structures. The envelope was created using the Density Plot module.
input files
output files
caution: DCD file is > 450 MB
hiv1_gag_monte_carlo.dcd
hiv1_gag_monte_carlo.dcd.all_rg_results_data.txt
hiv1_gag_monte_carlo.dcd.accepted_rg_results_data.txt
hiv1_gag_monte_carlo.dcd.stats
The input variables are listed below.
low Rg cutoff Structures with Rg values less than this value are discarded.
high Rg cutoff Structures with Rg values greater than this value are discarded.
check box to use Z coordinate filter Check box to implement the ability to discard structures with any Z coordinates with a value less than the user supplied Z cutoff value.
directed Monte Carlo (0==no or Rg value) Enter a non-zero value to use an extra energy term in the Monte Carlo sampling to favor Rg values towards the supplied value. The default value is zero which indicates that no bias is implemented.
check box to use atomic constraints Check box to implement the ability to discard structures that do not satisfy the atomic / geometric constraints provided in the user defined constraint file.
In the following sections examples will be shown for the various options in the Advanced Input section.
Advanced Example 1: Rg cutoffs
This example uses the low Rg and high Rg cutoff inputs to restrict accepted structures to be between 55 and 60. Note that the example reports the number and percent of Rg values that do not satisfy the input cutoffs.
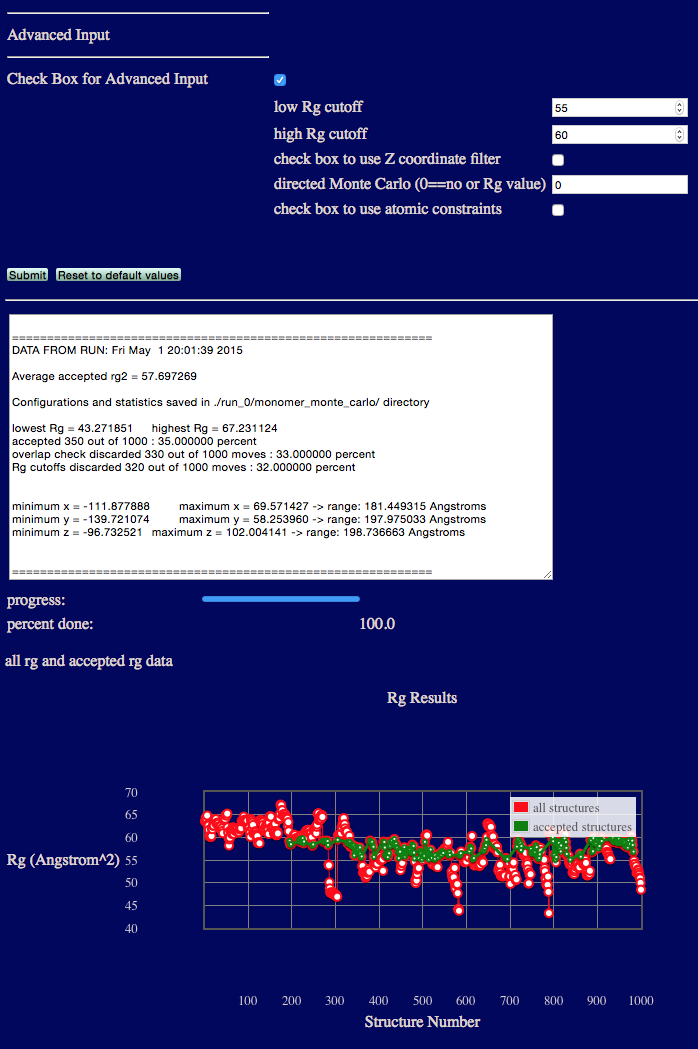
input files
output files
hiv1_gag_monte_carlo_rg_55_to_60.dcd
hiv1_gag_monte_carlo_rg_55_to_60.dcd.all_rg_results_data.txt
hiv1_gag_monte_carlo_rg_55_to_60.dcd.accepted_rg_results_data.txt
hiv1_gag_monte_carlo_rg_55_to_60.dcd.stats
Advanced Example 2: Z coordinate filter
This example restrict accepted structures to be those with all Z coordinates to be greater than 0.
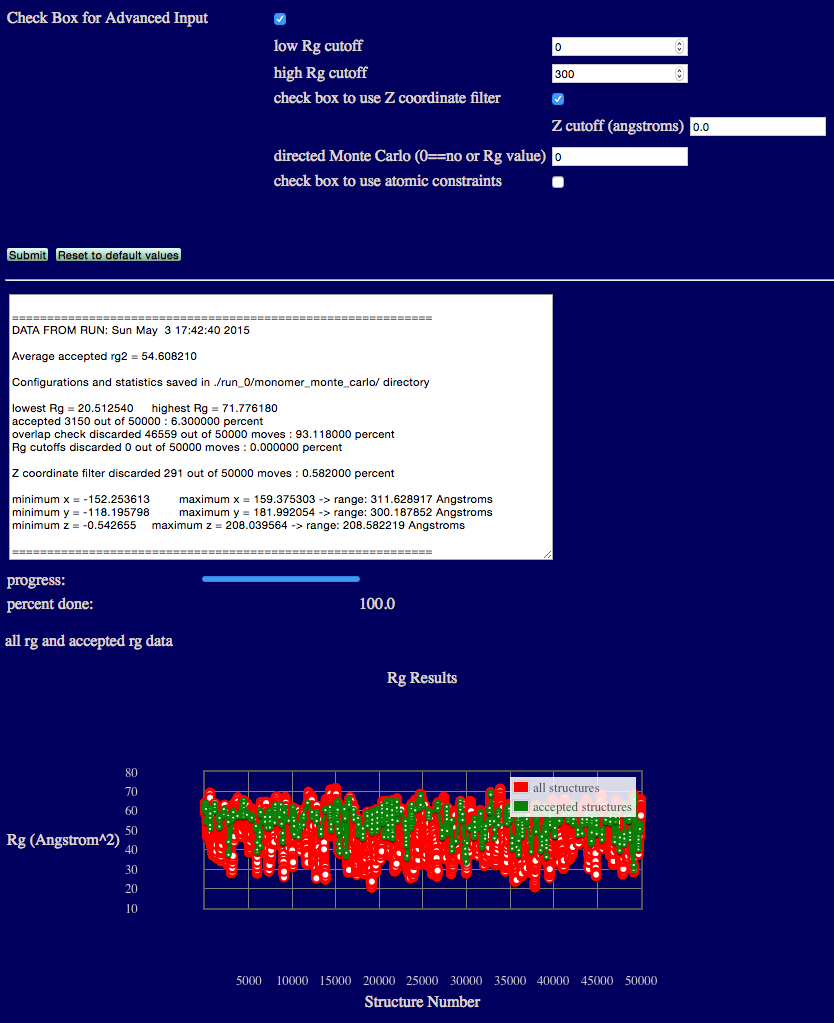
In the figure below, the original input structure of hiv1_gag inside the envelope sampled by all accepted structures. The envelope was created using the Density Plot module.
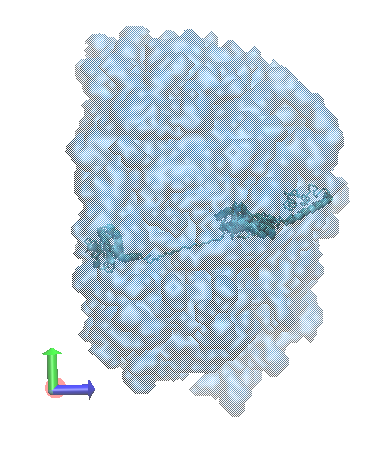
input files
output files
caution: DCD file is > 240 MB
hiv1_gag_on_membrane.dcd
hiv1_gag_on_membrane.dcd.all_rg_results_data.txt
hiv1_gag_on_membrane.dcd.accepted_rg_results_data.txt
hiv1_gag_on_membrane.dcd.stats
Advanced Example 3: Directed Monte Carlo
This example biases the Monte Carlo sampling to accept Rg values closer to 30.
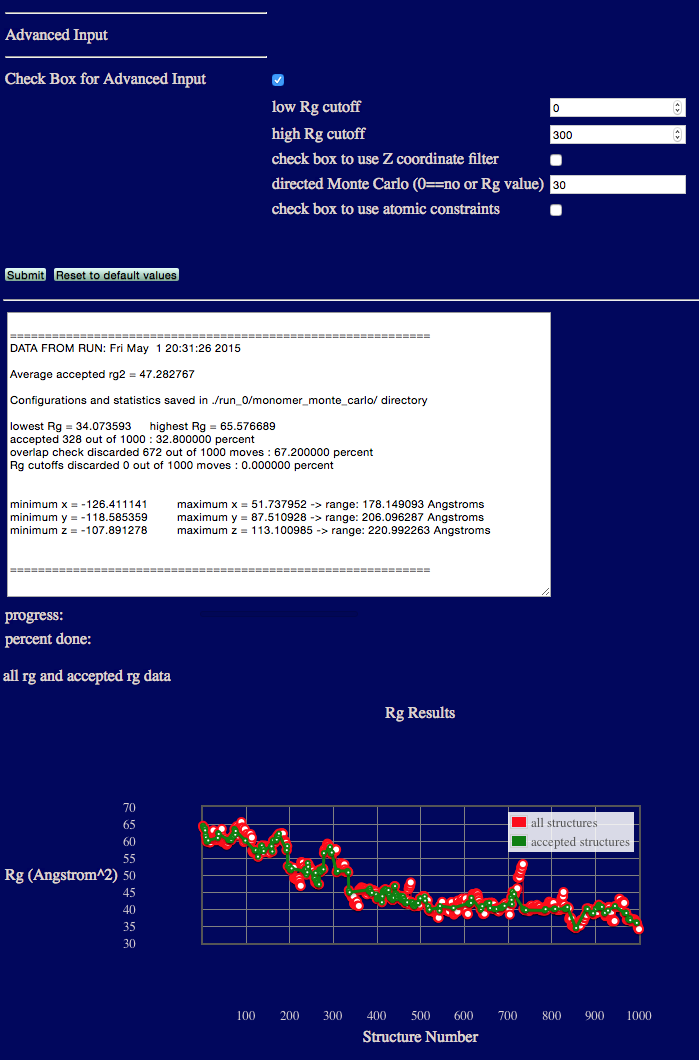
input files
output files
hiv1_gag_monte_carlo_directed_rg_30.dcd
hiv1_gag_monte_carlo_directed_rg_30.dcd.all_rg_results_data.txt
hiv1_gag_monte_carlo_directed_rg_30.dcd.accepted_rg_results_data.txt
hiv1_gag_monte_carlo_directed_rg_30.dcd.stats
Advanced Example 4: Atomic Constraints
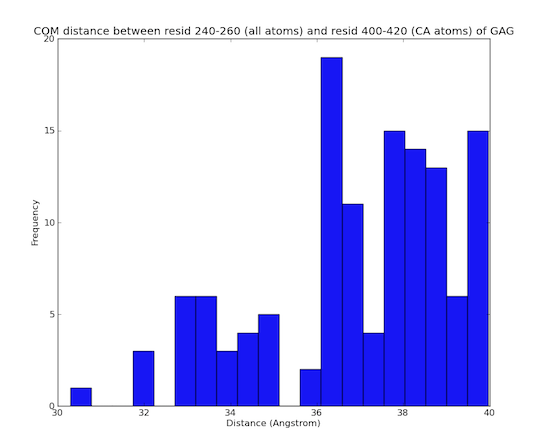
This example only accepts structures that satisfy the user defined atomic constraints. The segment name of the protein in the hiv1_gag.pdb is "GAG". The following single line, supplied in the user supplied file "constraints.txt" will filter the structures so that only structures with the center of mass of atoms in residues 240 to 260 is within 40.0 angstroms of the center of mass of CA atoms in residues 400 to 420.
Note that the constraint syntax is robust and allows for sophisticated selections, see Constraints for further details.
GAG 240-260 : GAG 400-420 CA : 40.0 : COM : COM
input files
output files
hiv1_gag_monte_carlo_constraints.dcd
hiv1_gag_monte_carlo_constraints.dcd.all_rg_results_data.txt
hiv1_gag_monte_carlo_constraints.dcd.accepted_rg_results_data.txt
hiv1_gag_monte_carlo_constraints.dcd.stats
In the figure below a plot of distances between the center of mass of residues 240 to 260 and the center of mass of CA atoms for residues 400-420 are shown for accepted structures from the simualtion utilizing constraints.
The program is written so that linear polymers of proteins and single-stranded nucleic acids are simulated over a specific selection of residues in a single direction.
A solution for the best rotation to relate two sets of vectors W. Kabsch, Acta Crystallog. sect. A 32 922-923 (1976). BIBTeX, EndNote, Plain Text
A discussion of the solution for the best rotation to relate two sets of vectors W. Kabsch, Acta Crystallog. sect. A 34 827-828 (1978). BIBTeX, EndNote, Plain Text
CHARMM: The energy function and its parameterization with an overview of the program A. D. MacKerel Jr., C. L. Brooks III, L. Nilsson, B. Roux, Y. Won, M. Karplus, The Encyclopedia of Computational Chemistry, John Wiley & Sons: Chichester, 271-277 (1998). BIBTex, Endnote, Plain Text
Conformation of the HIV-1 Gag Protein in Solution S. A. K. Datta, J. E. Curtis, W. Ratcliff, P. K. Clark, R. M. Crist, J. Lebowitz, S. Krueger, A. Rein, J. Mol. Biol. 365, 812-824 (2007). BIBTex, Endnote, Plain Text
SASSIE: A program to study intrinsically disordered biological molecules and macromolecular ensembles using experimental scattering restraints J. E. Curtis, S. Raghunandan, H. Nanda, S. Krueger, Comp. Phys. Comm. 183, 382-389 (2012). BIBTeX, EndNote, Plain Text