Generates a report that characterizes the user supplied PDB file.
The PDB Scan module is accessible from the Build section of the main menu.
The purpose of this module is to assess whether an input PDB is ready for simulation and where possible to provide files enabling CHARMM forecfield parameterization. When a header is available, two scans of an input PDB are made, one accounting for information extracted from header records and one based solely on information contained within the coordinate records. Both scans produce independent reports and in cases where a header is available this is deemed the most reliable guide to the files contents.
Information on missing atoms and residues and those not covered as standard by the CHARMM 27 forcefield are reported. Furthermore, when header information is available the BIOMT records are extracted to indicate if the protein unit in the coordinates is represenative of the biological unit or whether a coordinate transform is needed to complete it (e.g. in cases where symmetry can be used to create a dimer or higher order oligomer). Both header and no header reports provide information on (predicted) disulphide bonds found within the structure.
The text area report provides summary information on missing atoms and residues and those not covered as standard by the CHARMM 27 forcefield
All Header scan information is extracted from the PDB header records
Header scan checks for consistency between the SEQRES and coordinate sequences
No Header scan reports missing atoms by comparing coordinates to the CHARMM 27 residue descriptions
No Header scan detects possible missing residues by calculating the backbone C-N distance between two adjacent residue listed in the PDB coordinate entry.
No Header scan detects possible disulfide bonds by calculating the pair-wise distance between all Cys-S atoms.
These examples show the analysis and file output of a run scanning a PDB with and without header records. The former system requires editing before it can be prepared for simulation, the latter is ready to simulate.
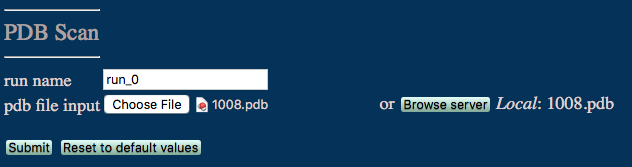
run name user defined name of folder that will contain the results.
pdb input file PDB file to be scanned for simulation suitability.
A brief summary report is shown in the textbox indicating whether the PDB is ready for simulation and that a full report has been successfully generated.
This example shows the report for a PDB containing a single chain in the coordinates but which the header indicates has a dimeric biological unit.
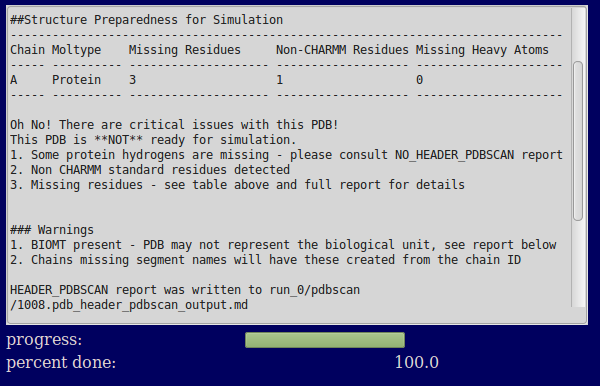
The text output region provides a brief summary of the contents of the file. It focusses on issues that might prevent the creation of a correct CHARMM parameterized system (missing residues and atoms and unusual residues not standard in CHARMM). If the PDB is not ready for simulation (as is the case in this example) many of the lines point to the full report for more details.
A full report is provided which details all of the information extracted from the PDB header and coordinates. The first part of the report reproduces the text area report for future reference.

The next section of the gives information on the totality of the contents of the PDB.
The summary section of the main report gives the high level information on the protein provided in the PDB header relating to what it represents and the origin of the data.
The polymers section gives information on the residues in each polymer chain in the file.
The heterogens section describes all of the unique non-standard residues reported in the header

The system statistics section provides basic information on the geometry and mass of the protein calculated from the atomic coordinates. A similar table is also provided per chain later in the report.
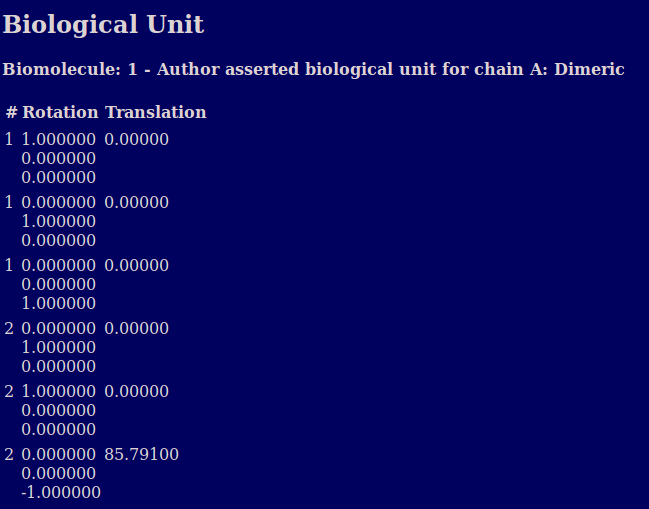
The biological unit section describes the oligomeric state suggested by the authors of the PDB (and/or the software they used) in the BIOMT records.. It also shows the geometric transformation matrix provided in the PDB header to transform the provided coordinates where multiple copies are needed to produce the biological unit.
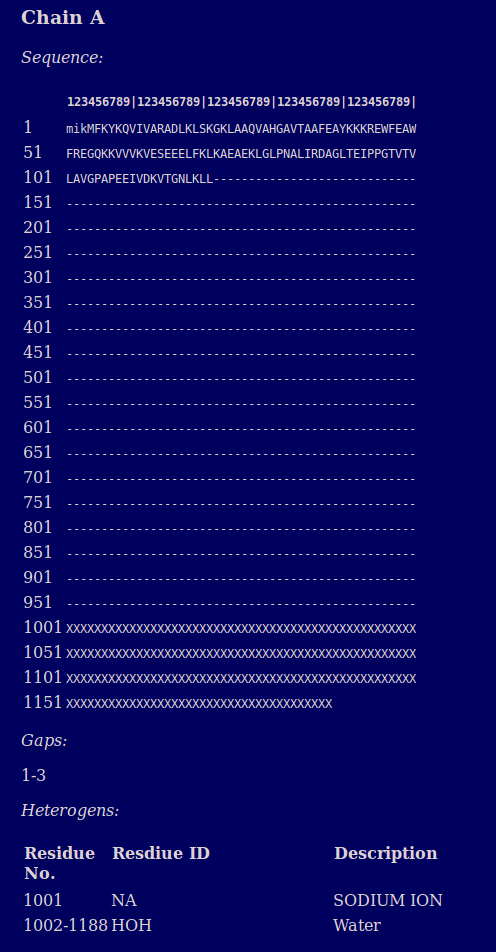
A report is provided for each chain which reports on the sequence in FASTA format and any heterogens present. Lower case letters in the sequence indicate missing residues
In addition to producing a report detailing the contents of the files two PDB files are generated for each chain. The first contains full entries including the coordinates of all the atoms present in the input PDB. the second a single CA atoms for each residue in the full sequence described in the header (whether or not coordinates were found in the input PDB). These files are used in PSFGEN to create completed PDB/PSF pairs for simulation.
As part of the outpt a JSmol vizualization of the protein is produced. Holding down the left mouse button and moving the cursor over the picture allows you to rotate the view, the scroll wheel facilitates zooming in and out. Right clicking on the image allows you to access all of the JSmol options.

input files
output files
1008.pdb_header_pdbscan_output.html
1008.pdb_header_pdbscan_output.md
1008.pdb_no_header_pdbscan_output.html
This example shows the report for a PDB that contains no header.
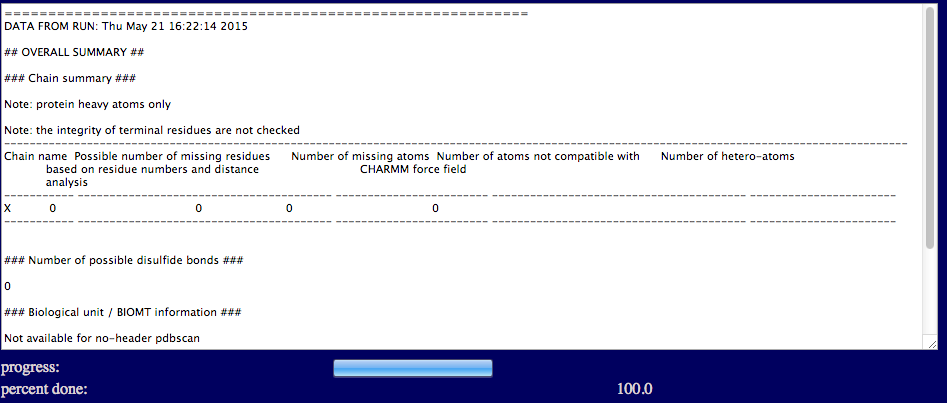
The text output region provides a brief summary of no header PDBScan report, that include the number of missing residues, missing atoms, and hetero-atoms for each individual chain, and the number of disulfide bonds. The number of missing atoms and the number of disulfide bonds are given based on distance analysis as described in the previous section.
A full report is provided which details all of the information extracted from the PDB coordinate entry.
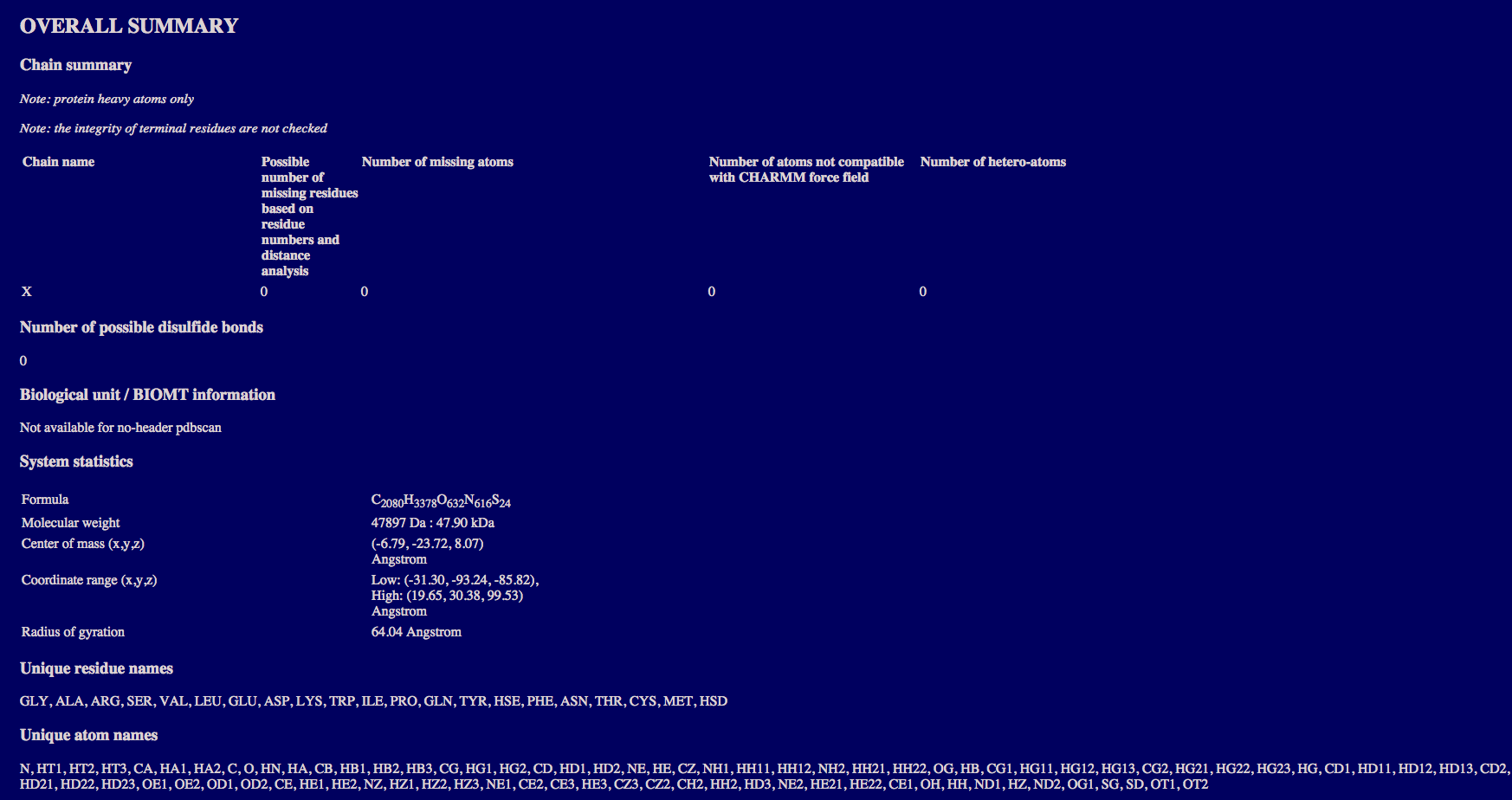
The overall summary section provide:
the number of missing residues, missing atoms, and hetero-atoms for each individual chain;
the number of disulfide bonds;
the system statistics including the formula, molecular weight, and geometric statistics;
and the unique residue names and atom names present in the coordinate entry.
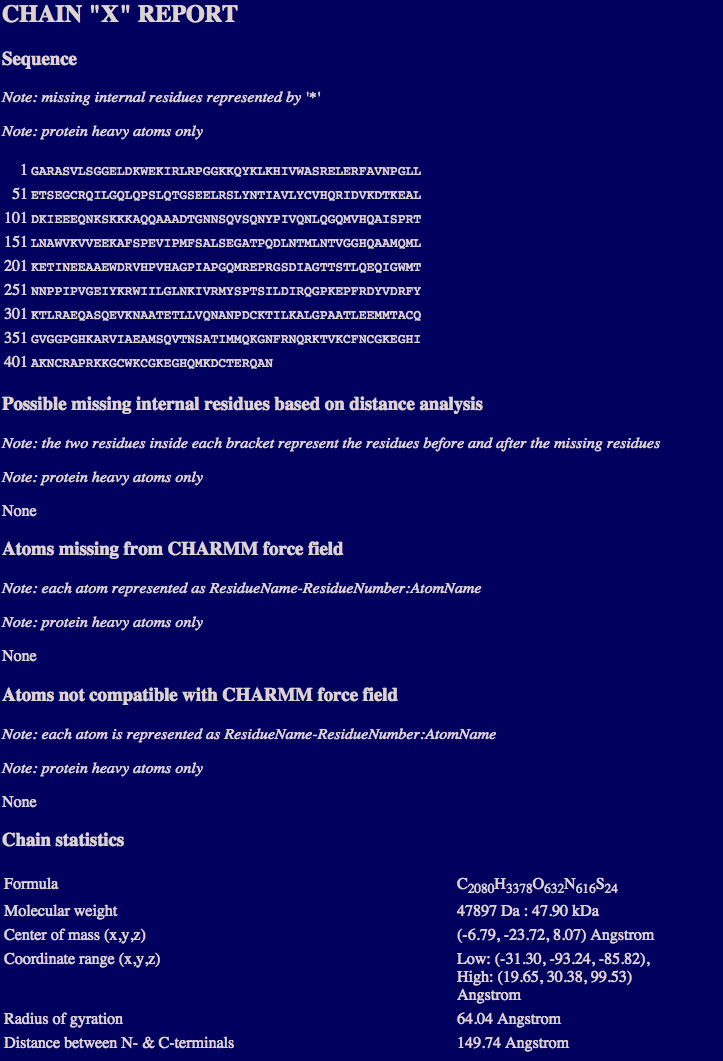
The chain report section may contain multiple sections, each of which provides for the individul chain:
the sequence;
the number of missing regions based on distance analysis;
the number of missing atoms from the CHARMM force field;
the number of atoms that are not compatible with CHARMM force field;
and the chain statistics including formula, molecular weight, and geometric statistics.

The hetero-atom section lists the hetero-atoms that are compatible and not compatible with CHARMM force field.

The disulfide bond section lists the disulfide bonds that are found by distance analysis.
As part of the outpt a JSmol vizualization of the protein is produced. Holding down the left mouse button and moving the cursor over the picture allows you to rotate the view, the scroll wheel facilitates zooming in and out. Right clicking on the image allows you to access all of the JSmol options.
input files
output files