 \
\Performs energy minimization and molecular dynamics simulation of input structures.
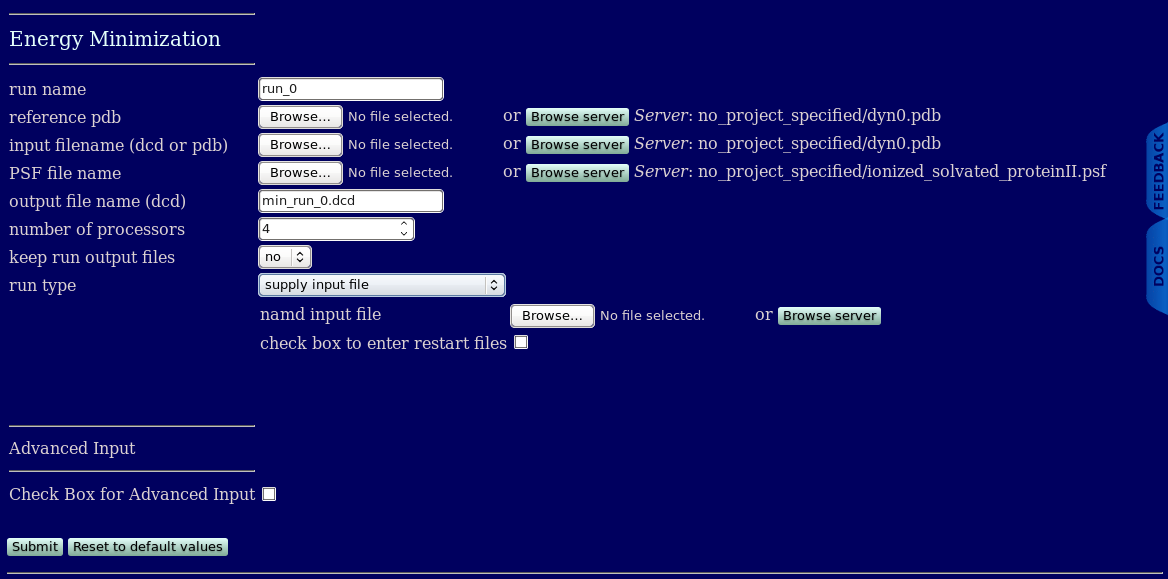
The Energy Minimization module is accessible from the Simulate section of the main menu.
The purpose of the module is to use energy minimization and molecular dynamics to remove bad contacts from biomolecular models parameterized in the CHARMM forcefield.
Users will input a reference PDB file name, along with matching starting structure (in either PDB or DCD format) and CHARMM topology (PSF) files. Four modes of operation can be chosen:
minimization alone
minimization followed by molecular dynamics
minimization followed by molecular dynamics leading to a second round of minimization
molecular simulation (energy minimization and/or molecular dynamics) with a user supplied input file.
These four options are explored in three cases below.
NAMD (version 2.9) is used as the simulation engine
Both minimization and molecular dynamics are performed using the Generalized Born implicit solvent model
If a DCD file is selected as the input file then simulations are run on each frame
These examples show minimization of a single structure and a minimization and molecular dynamics run using advanced inputs from a DCD containing several structures.
This example minimizes a single structure input as a PDB using the default CHARMM 27 forcefield.
\
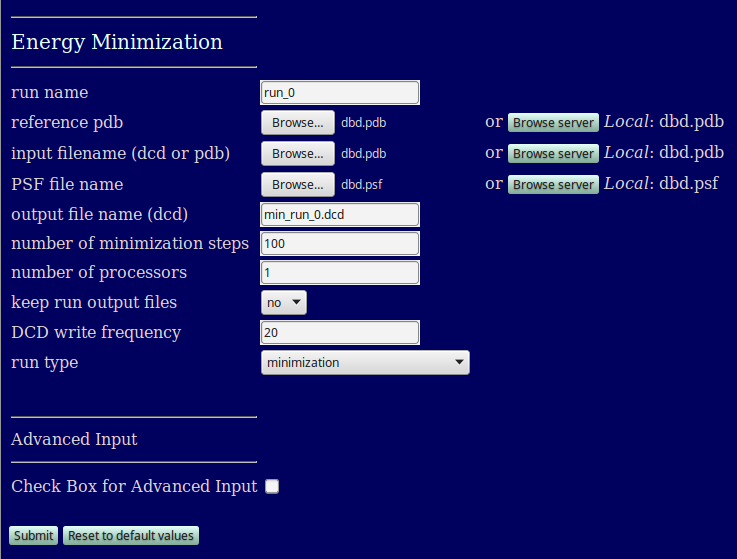
run name: user defined name of folder that will contain the results.
reference pdb: PDB file with naming information for coordinates that will be extracted.
input filename (dcd or pdb): file containing starting conformation(s) for simulation. The number of atoms must match that in the reference pdb. For files with multiple frames each one will be simulated.
PSF file name (dcd or pdb): PSF file with topology information, must match the reference pdb and input dcd/pdb
output file name (dcd): filename for the output DCD contaiing the final frames resulting from simulation
number of minimization steps: number of steps of the conjugate gradient minimization to apply to each structure
number of processors: number of processors used to run the simulation
keep run output files: choice of whether to retain NAMD log files and other output from each simulation
DCD write frequency: user determined number of steps between the output of structures from each simulation
run type: select which of the four combinations of minimization and molecular dynamics to run
 \
\
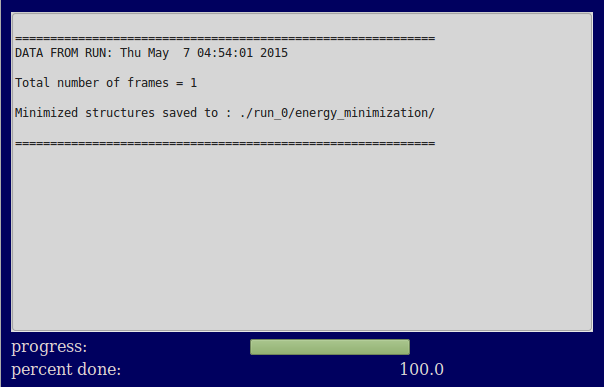
./run_0/minimization/dbd.pdb
./run_0/minimization/dbd.psf
./run_0/minimization/min_run_0.dcd
./run_0/minimization/min_run_0.dcd.pdb
Input files
Output files
This example minimizes and then runs moleclar dynamics on every structure in the input DCD using the CHARMM 36 forcefield.
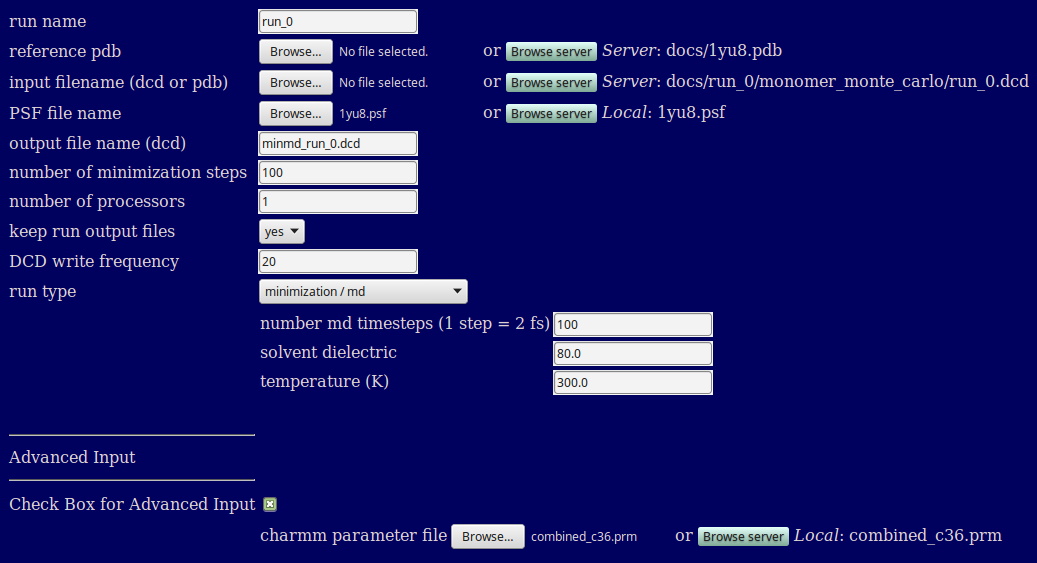
When a run type of minimization/md or minimization/md/minimization are selected additional options related to the molecular dynamics simulation are revealed.
number md timesteps (1 step = 2 fs): number of molecular dynamics steps to be run if selected as part of the run type (ignored for minimization alone runs)
solvent dielectric: Defines the dielectric of the implicit solvent used in teh simulation, usually 78.5 or 80 (the latter of which is the default).
temperature (K): temperature in Kelvin at which simulatons will be performed
The advanced input section is accessed by ticking the "Check Box for Advanced Input".
 \
\
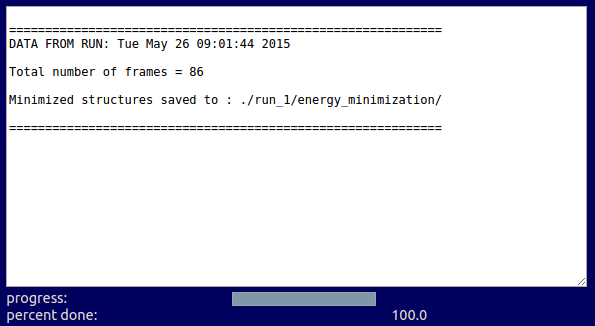
./run_0/minimization/1yu8.pdb
./run_0/minimization/1yu8.psf
./run_0/minimization/minmd_run_0.dcd
./run_0/minimization/minmd_run_0.dcd.pdb
min_00001.out
...
min_000086.out
temp.inp
Input files
Output files
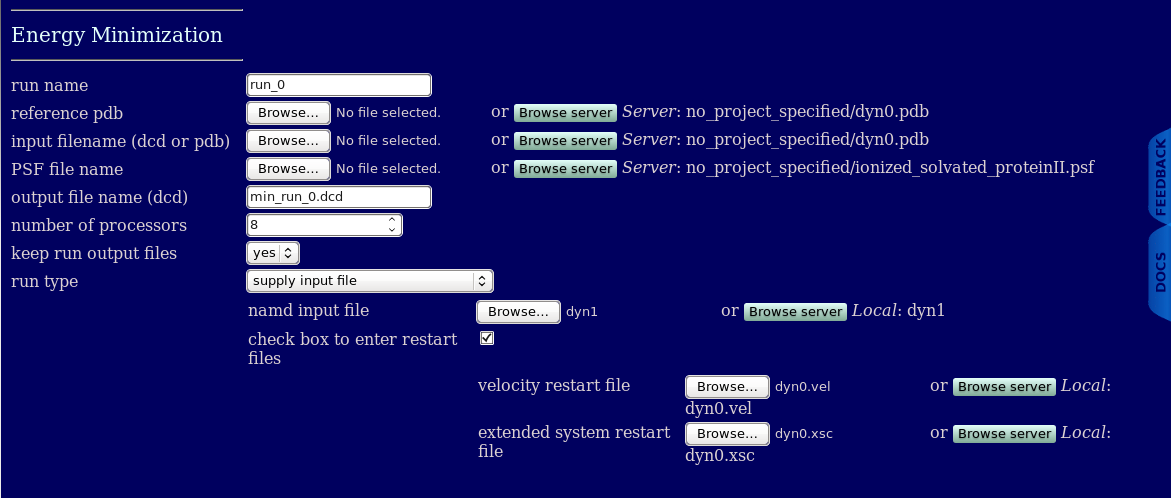
It is possible to provide an input file that allows for the simulation of more complicated systems.
This aspect is for experienced users.
When a run type of supply input file is chosen then one can select the name of the namd input file name.
The PDB, (DCD/PDB), PSF, and output file name read in from SASSIE-web take precendence for the files that may be listed in the user supplied input files. Specifically, the following input file keywords are overridden by the values provided by the userinput in SASSIE-web
coordinates
structure
paratypecharmm
parameters
outputname
DCDfile
It is also possible to upload velocity and extended system files which is often required for continuuing / restarting a previous trajectory.
When a run type of supply input file is chosen and one selects the check box to enter restart files then the option to enter the filename(s) of velocity restart file and/or extended system restart file is provided.
If you check the box to enter restart files then the following keywords are overridden by the values provided by the user in SASSIE-web
velocities
extendedSystem
NOTES
restartname output/dyn0.rest.pdb
should be written as
restartname dyn0.rest.pdb
Input files
Output files
[]
Scalable molecular dynamics with NAMD James C. Phillips, Rosemary Braun, Wei Wang, James Gumbart, Emad Tajkhorshid, Elizabeth Villa, Christophe Chipot, Robert D. Skeel, Laxmikant Kale, and Klaus Schulten. J. Comput. Chem. 26, 1781-1802 (2005). BIBTeX EndNote Plain Text
SASSIE: A program to study intrinsically disordered biological molecules and macromolecular ensembles using experimental scattering restraints J. E. Curtis, S. Raghunandan, H. Nanda, S. Krueger, Comp. Phys. Comm. 183, 382-389 (2012). BIBTeX EndNote Plain Text