Generates structures by rigid body translations and rotations of one structure about a second structure.
The Two-Body Grid module is accessible from the Simulate section of the main menu.
The purpose of the module is to perform rigid body displacements of an input structure (molecule 2) about a second (fixed) structure (molecule 1). Resultant structures contain both molecules.
The starting structures must have residue fields at a minimum. However, if this module is to be used to generate structures to compare to experimental data using SasCalc, the starting structures must be complete, i.e, without missing residues. Atom and residue naming must be compatiable with those defined in the CHARMM force field See Notes on Starting Structures and Force Fields and PDB Scan for further details.
The output file format is DCD since in most cases many structures are generated. There is no option to save the output files in PDB format. One can use Extract Utilities to convert DCD files to multi-frame PDB files.
Atomic overlap is checked using the CA atoms and a distance cutoff value of 3.0 A. A newly-generated structure is accepted only if there is no overlap between CA atoms.
In Advanced Input, options are provided to reject structures based on Rg value, position of atoms in the Z-direction and via atomic constraints provided as a list in a text file as described in Constraints. These options are not mutually exclusive and can be used in the same run as needed.
Typical workflows involve generating an ensemble of structures using this module, then energy minimizing the ensemble using Energy Minimization, then calculating scattering from the ensemble using modules in Calculate, and finaly comparing results to experimental data using modules in Analyze.
CAUTION: The output DCD files can become large quickly depending on the number of translations and rotations that are performed and the number of structures that are accepted. It is advisable to perform coarse moves initially and compare the resultant structure to experimental data to narrow down the region(s) of interest for molecule (2). Then, finer moves can be made to refine the structure.
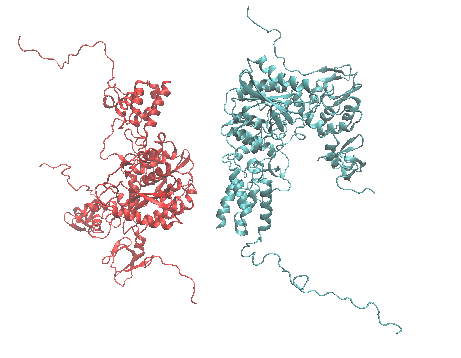
This example generate stuctures of the HIVintegrase/LEDGF complex. One half of the complex (red) consists of two HIVintegrase subunits and one LEDGF subunit and serves as the fixed reference structure. The second half of the complex (blue) is translated and rotated about the first half to form structures of the entire complex.
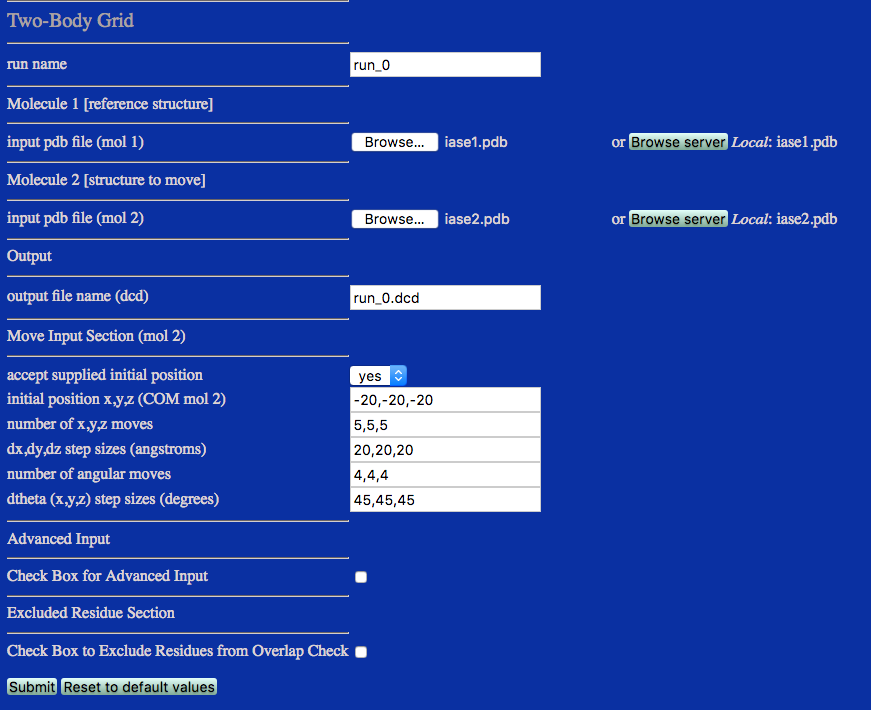
run name User defined name of folder that will contain the results.
input pdb file (mol 1) PDB file for molecule 1 as reference structure.
input pdb file (mol 2) PDB file for molecule 2 that moves about molecule 1.
outout file (dcd) Name of DCD file with the new coordinates for molecule 2.
Move Input Section (mol 2)
accept supplied initial position Select Yes to set the center of mass (COM) of molecule 2 to a specific position. If No is selected the COM of molecule 2 will be calculated from the coordinates in the input PDB file.
initial position x,y,x (COM mol 2) Position of COM for molecule 2 (will be used if above choice is Yes).
number of x,y,z moves Number of moves for each cartesian coordinate.
dx,dy,dz step sizes (angstroms) Distance between subsequent translations of the COM of molecule 2.
number of angular moves Number of rotational moves for each Euler angle.
dtheta (x,y,z) step sizes (degrees) Angular distances between each subsequent rotation about COM of molecule 2.
./run_0/two_body_grid/run_0.dcd.pdb
./run_0/two_body_grid/run_0.dcd
./run_0/two_body_grid/gridrun_output.txtrun_0.dcd.pdb contains the intial coordinates for the complex containing both molecule 1 and 2.
run_0.dcd contains trajectories of the entire complex in which molecule 2 is displaced around molecule 1 as defined in the Move Input Section.
grid_output.txt contains the COM positions (x,y,z) and angular values (thetax, thetay, thetaz) of molecule 2 for the accepted structures.

Check Box for Advanced Input Click for extra options.
low Rg cutoff Discard all accepted structures with a Rg less than this value.
high Rg curoff Discard all accepted structures with a Rg greater than this value.
Z coordinate filter Choose yes to discard all accepted structures with any atomic coordinate (in the z-axis) less than Z cutoff value defined below.
Z curoff (angstroms) Discard all accepted structures with any atomic z coordinate less than this value.
check box to use atomic constraints Check if you wish to enable atomic constraints.

Check Box to Exclude Residues from OVerlap Check Check if you wish to EXCLUDE specific atoms in the check for atomic overlap. This is useful if molecule 1 or molecule 2 already has some overlaping residues. These residues can be excluded from the overlap check that is made after each move to determine if the new structure will be accepted.
number of segments with excluded residues (mol 1) Number of segments with residues to omit from overlap check (mol 1).
name of segments with excluded residues (mol 1) Name(s) of segments with residues to omit from overlap check (mol 1).
first amino acid per segment (mol1) Number of first residue in each of the excluded segments (mol 1).
number of contiguous amino acids per segment (mol 1) Number of the contiguous residue(s) in each of the excluded segments (mol 1)
number of segments with excluded residue (mol 2) Number of segments with residues to omit from overlap check (mol 2).
name of segments with excluded residues (mol 2) Name(s) of segments with residues to omit from overlap check (mol 2).
first amino acid per segment (mol 2) Number of first residue in each of the excluded segments (mol 2).
number of contiguous amino acids per segment (mol 2) Number of the contiguous residue(s) in each of the excluded segments (mol 2)
input files
output files
SASSIE: A program to study intrinsically disordered biological molecules and macromolecular ensembles using experimental scattering restraints
J. E. Curtis, S. Raghunandan, H. Nanda, S. Krueger
Comp. Phys. Comm. 183, 382-389 (2012).
BIBTeX Citation
EndNote Citation
Plain Text Citation