Calculates neutron scattering profiles of proteins and nucleic acids from input structures.
The Cryson module is accessible from the Retired section of the main menu.
The purpose of the module is calculate neutron scattering profiles from a user supplied structure file. It is configured to handle trajectory input files (DCD or PDB).
Complete usage notes can be found at the EMBL site for Cryson.
Module calculates neutron scattering profiles for proteins.
The default input file format is DCD. For large numbers of frames we recommend saving your data in the DCD format (~seven fold smaller file size).
The program does not deterine if the input parameters are adequate to converge the scattering profile over the desired range of q. This is the responsbilitiy of the user.
Even if only one structure is to be used to calculate a theoretical scttering curve, it still needs a reference structure even if the coordinates are in a PDB file.
More data points may not be mathematically justified. For SAS there is limited information content related to the size of the molecule measured in the experimental scattering data. 15 to 31 points are generally used. See BIOISIS for a theoretical and pratical reasoning regarding the number of points one should use.
The number of q-points, range of q, and the spacing of the q-points used to create interpolated data files MUST match the input settings that you use in this module to calculate SANS profiles if you wish to compare theoretical and experimental data (Chi-Square Filter).
The final range of q will be from 0 to a maximum q-value of (number of q-values - 1) * delta q, where delta q is the spacing between adjacent q-values entered by the user.
The calculated SANS profiles can be extracted using Extract Utilities or combined from multiple runs using Merge Utilities.
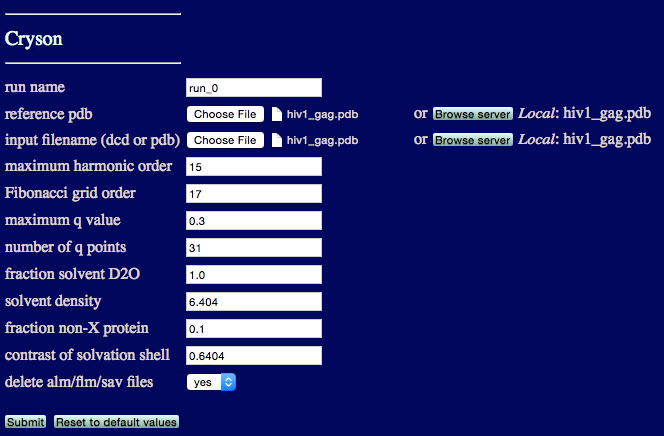
This example calcualtes a SANS profile for hiv1 GAG protein.
run name: user defined name of folder that will contain the results.
reference pdb: PDB file with naming information for coordinates that will be extracted
input filename (dcd or pdb): DCD or PDB file with coordinates that will be used to calculate scattering profile(s).
maximum harmonic order Choose enough basis functions for converged scattering profile(s).
Fibonacci grid order Choose enough basis functions for converged scattering profile(s)
maximum q value: The maximum value to calculate I(q).
number of q points The number of individual q-points, including I(0).
fraction solvent D2O Enter a decimal number between 0 and 1.
solvent denisty Enter appropriate value.
fraction non-X protein Enter appropriate value.
contrast of solvation shell Enter appropriate value.
delete alm/flm/sav files: Option to delete additional files created for each structure.
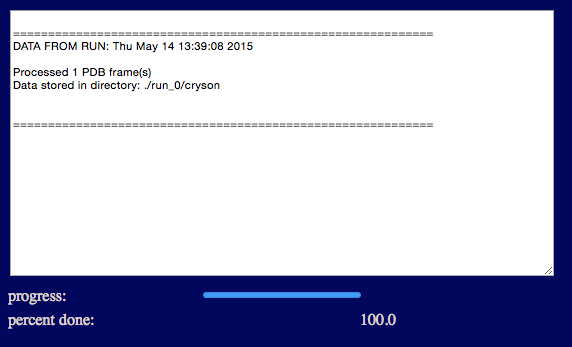
The output will indicate the number of processed frames and the location of the scattering profiles.
Results will be written to a new directory within the given "run name". For example, in the figure it is noted that the structures and dimensions were saved files within the current project directory within the chosen "run name" directory:
./run_0/cryson/
None
input files
output files
scattering profile and associated files are provided as a compressed archive
Source code is not accessible to troubleshoot usage and physical details of the calculator.
Protein hydration in solution: experimental observation by X-ray and neutron scattering D. I. Svergun, S. Richard, M. H. J. Koch, Z. Sayers, S. Kuprin, G. Zaccai, Proc. Natl. Acad. Sci. USA 95, 2267-2272 (1998). BIBTeX, EndNote, Plain Text
SASSIE: A program to study intrinsically disordered biological molecules and macromolecular ensembles using experimental scattering restraints J. E. Curtis, S. Raghunandan, H. Nanda, S. Krueger, Comp. Phys. Comm. 183, 382-389 (2012). BIBTeX, EndNote, Plain Text